
新藥從研發到上市的全流程:IND、NDA、ANDA傻傻分不清?
發布時間:2023-03-01來源:點擊:1019
以下文章來源于:小藥說藥
一款新藥從研發到上市都需要經過哪些流程?每一步又有哪些經驗可以借鑒?
臨床前研究1
研究開發(一般 2-3年)
實驗室研究,尋找治療特定疾病的具有潛力的新化合物。
- 藥物靶點的發現及確認
這是所有工作的起點,只有確定了靶點,后續所有的工作才有展開的依據。
- 化合物的篩選與合成
根據靶點的空間結構,從虛擬化合物庫中篩選一系列可匹配的分子結構,合成這些化合物,它們被稱為先導化合物。
- 活性化合物的驗證與優化
不是所有先導化合物都能符合要求,在這個階段需要通過體外細胞試驗驗證,初步篩選出活性高、毒性低的化合物,并根據構效關系進行結構優化,這些化合物稱為藥物候選物。
同時也存在一個化合物對目標 A 靶點沒有作用,卻有可能對其他的 B 靶點、C 靶點有非常好活性的情況,暫且不表。
1 臨床前實驗(一般 2-4年)
這一階段目的,一是評估藥物的藥理和毒理作用,藥物的吸收、分布、代謝和排泄情況(ADME)。二是進行生產工藝、質量控制、穩定性等研究(CMC)。
第一部分的實驗需要在動物層面展開,細胞實驗的結果和活體動物實驗的結果有時候會有很大的差異。這一步的目的是確定藥物的有效性與安全性。
第二部分需要在符合GMP要求的車間完成。
- 藥理學研究
包括:藥效學、藥動學
- 毒理學研究
急毒、長毒、生殖毒性,致癌、致畸、致突變情況
- 制劑的開發
總不能弄點化合物就直接往嘴里倒吧,制劑開發是藥物應用的一個重要環節。比如有的藥口服吸收很差,就需要開發為注射劑。
有的藥在胃酸里面會失去活性,就需要開發為腸溶制劑。
有的化合物溶解性不好,這也可以通過制劑來部分解決這個問題。
還有的需要局部給藥,就需要通過制劑開發成霧化劑、膏劑等。
2 臨床試驗審批(IND)

3 臨床試驗(一般3-7年)
人體試驗共分三期:
- Ⅰ期臨床 20-100例,正常人,主要進行安全性評價。
- Ⅱ期臨床 100-300例,病人,主要進行有效性評價 。
- Ⅲ期臨床 300-5000例,病人,擴大樣本量,進一步評價。
因為Ⅰ-Ⅲ 期臨床在整個藥物研發過程中非常重要,我們重點講一下這部分。
傳統意義上,新藥的臨床研究分為I/II/III期,后來 II 期又分成 IIa 和 IIb(很大程度上是因為腫瘤藥物研究),接著出現了 0 期研究的概念(很大程度上也是因為腫瘤藥物的研發)。然后又有人提出,0/I 期為早期臨床研究,IIa 為中期臨床研究,IIb 和 III 期為晚期臨床研究。
這種分類,其實我覺得在兩個方面對我們進行臨床研究設計的人來說,是很重要的:
1. 分期是和研究目的相關的,不同的目的,可以分到不同時期/階段的。
2. 不同時期/階段的臨床研究,往往對應于一定的研究框架設計(交叉研究,平行對照,單臂,終點選擇等等)。
所以,理解不同時期/階段的臨床研究,其意義已經超出了知道有這樣的分法本身。
分期的一頭是研究目的,另一頭是對應的設計框架(當然也只是粗略的框架,很多問題,要根據疾病、藥物等來確定),理解分期,有助于我們了解,一個研究目的,大體上有哪些設計方法和思路,大體上可以如何去處理;為什么該如此進行,各種設計的優缺點在哪里。
所以,我們先看一下不同階段的研究目的。
1.0 期
目的是在滿足一定的統計學要求的前提下,在有限的樣本量(通常不超過20)、有限的時間內(每個患者的治療期常不超過數周),初步判斷一個研究藥物是否有效、某個劑量是否有效,是否應繼續開發下去,附帶看一下某些療效判斷方法是否可行。
從目的角度,0 期研究也是初步判斷藥物的效果,類似 II 期(尤其是IIa),所以 0 期的設計,在某些方面類似 IIa 的單臂研究,但是由于樣本量小、又要滿足一定的統計學要求,所以也有其特點,設計起來其實可能更為復雜。
2.I 期
通常是摸索劑量(dose finding, dose-ranging),藥代(PK)和藥動(PD),樣本量也不大(一般不超過20,或者20左右)。尤其是PD,通常會使用交叉研究,因為交叉研究有其特點,適合此類設計目的。
交叉研究中,每個患者都是其自己的對照(在不同階段,分別扮演研究組和對照組的角色),而且交叉研究的變量一般小于平行對照,所以在利用樣本方面,交叉研究的效率遠高于平行對照,交叉研究如果納入 20 例患者,平行對照 50 例都不一定能獲得相似的結果。
但是交叉對照也有很多限制,例如脫落和失訪對交叉研究的影響遠大于平行對照研究;同一個人,不同時期接受不同治療、中間隔以洗脫期,導致后遺效應,以及治療-時間相互作用等等。所以在后期臨床研發中,很少看到有用交叉對照的(有,但遠少于平行對照)。
3.II 期
II 期是初步判斷療效的和安全性的(其實安全性貫穿研發始終),所以一般稱為safety & activity研究(SA研究)。有多種做法,根據不同的臨床疾病環境,以及既往數據等,可以采取單臂,平行對照的 RCT 等,終點一般選擇 surrogate,而不是 clinical outcome。如果分 IIa 和 IIb,則 IIa 往往是單臂,樣本量一般不會過百;IIb 則往往是平行 RCT。
4.III 期
III 期是嚴格的驗證藥物效果的驗證性臨床研究。很多會議上介紹臨床研究,往往以此類研究為模版進行介紹,在假設檢驗的框架下進行介紹。
4 新藥上市審批
- NDA申報資料 — CTD(Common Technical Document)
CTD主要由五大模塊組成:
①行政和法規信息
②概述:藥物質量、非臨床、臨床試驗的高度概括
③藥品質量詳述
④非臨床研究報告
⑤臨床研究報告
流程:
①批準信
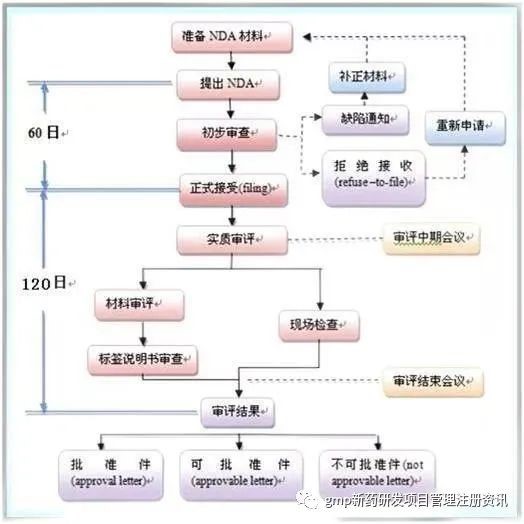
符合要求,可以上市
②可批準信
基本滿足要求,少數不足可以修改 。
申請人應在收到10日內作出回應修正,否則視為自動撤回。
③拒絕信
存在嚴重問題或需要補充大量信息資料。
申請人可在10日內提出修正或在30日內要求聽證。
- NDA特殊審評程序
①優先審評(Priority Reviews)
適用于能夠在治療、診斷或預防疾病上比已上市藥品有顯著改進的藥品,優先安排NDA審評。
②加速審批(Accelerated Approval)
用于治療嚴重或危及生命疾病的藥品,且存在合理并能夠測量的“替代終點”(Surrogate endpoint),即藥物預期的治療效果的指標,變通審評標準,利用“替代終點”審評。
③快速通道(Fast-track)
用于治療嚴重或危及生命疾病的藥品,且有潛力滿足臨床尚未滿足的醫學需求,早期介入,密切交流,分階段提交申報資料。
以萬絡為例,1998年11月23日萬絡提交NDA 申請,編號 21-042,“1999 年 5 月 20 日獲 FDA 批準,歷時 178 天。
5 上市后研究
臨床監測期:IV期臨床
受試者要大于 2000 例,同時要進行社會性考察。
仍以萬絡為例:2000 年進行了“VIGOR”胃腸道試驗 ——顯示較少的胃腸道副作用, 但是使用 18 個月后會引發 2 倍的心臟病/中風風險。
2001 年,“APPROVe”腺瘤息肉預防試驗 ——服藥超過 18 個月出現較高的心血管疾病風險。
6 上市后再審批(一般上市后4-10年)
目的:重新審核 NDA 中的有效性和安全性。

來源:小藥說藥

